Recording computational steps
Objectives
Understand why and when a workflow management tool can be useful
Questions
You have some steps that need to be run to do your work. How do you actually run them? Does it rely on your own memory and work, or is it reproducible? How do you communicate the steps for future you and others?
How can we create a reproducible workflow?
Instructor note
5 min teaching
15 min exercise/demo
Several steps from input data to result
The following material is partly derived from a HPC Carpentry lesson.
In this episode, we will use an example project which finds most frequent words in books and plots the result from those statistics. In this example we wish to:
Analyze word frequencies using code/count.py for 4 books (they are all in the data directory).
Plot a histogram using plot/plot.py.
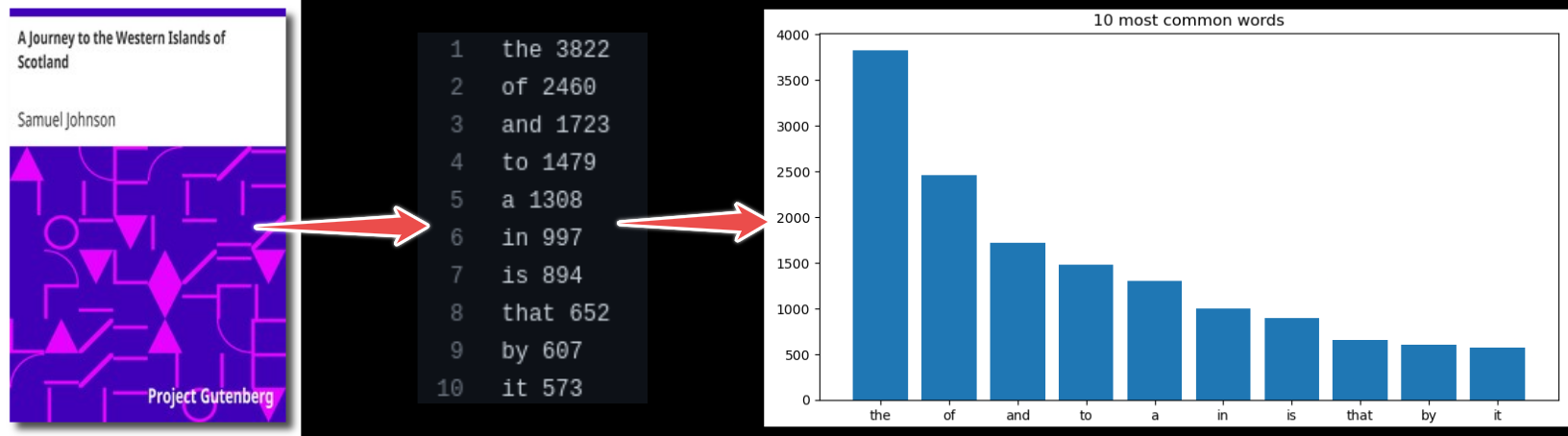
Example (for one book only):
$ python code/count.py data/isles.txt > statistics/isles.data
$ python code/plot.py --data-file statistics/isles.data --plot-file plot/isles.png
Another way to analyze the data would be via a graphical user interface (GUI), where you can for example drag and drop files and click buttons to do the different processing steps.
Both of the above (single line commands and simple graphical interfaces) are tricky in terms of reproducibility. We currently have two steps and 4 books. But imagine having 4 steps and 500 books. How could we deal with this?
As a first idea we could express the workflow with a script. The repository includes such script called run_all.sh.
We can run it with:
$ bash run_all.sh
This is imperative style: we tell the script to run these steps in precisely this order, as we would run them manually, one after another.
Discussion
What are the advantages of this solution compared to processing all one by one?
Is the scripted solution reproducible?
Imagine adding more steps to the analysis and imagine the steps being time consuming. What problems do you anticipate with a scripted solution?
Solution
The advantage of this solution compared to processing one by one is more automation: We can generate all. This is not only easier, it is also less error-prone.
Yes, the scripted solution can be reproducible. But could you easily run it, e.g., on a Windows computer?
If we had more steps and once steps start to be time-consuming, a limitation of a scripted solution is that it tries to run all steps always. Rerunning only part of the steps or only part of the input data requires us to outcomment or change lines in our script in between runs which can again become tedious and error-prone.
Workflow tools
Sometimes it may be helpful to go from imperative to declarative style. Rather than saying “do this and then that” we describe dependencies between steps, but we let the tool figure out the order of steps to produce results.
Example workflow tool: Snakemake
Snakemake (inspired by GNU Make) is one of many tools to create reproducible and scalable data analysis workflows. Workflows are described via a human readable, Python based language. Snakemake workflows scale seamlessly from laptop to cluster or cloud, without the need to modify the workflow definition.
A demo
Preparation
The exercise (below) and pre-exercise discussion uses the word-count repository (https://github.com/coderefinery/word-count) which we need to clone to work on it.
If you want to do this exercise on your own, you can do so either on your own computer (follow the instructions in the bottom right panel on the CodeRefinery installation instruction page), or the Binder cloud service:
On your own computer:
Install the necessary tools
Activate the CodeRefinery Conda environment with
conda activate coderefinery.Clone the word-count repository:
$ git clone https://github.com/coderefinery/word-count.git
On Binder:
We can also use the cloud service Binder to make sure we all have the same computing environment. This is interesting from a reproducible research point of view and it’s explained further in the Jupyter lesson how this is even possible.
Go to https://github.com/coderefinery/word-count and click on the “launch binder” badge in the README.
Once it gets started, you can open a new Terminal from the Launcher or via File > New > Terminal.
Workflow-1: Workflow solution using Snakemake
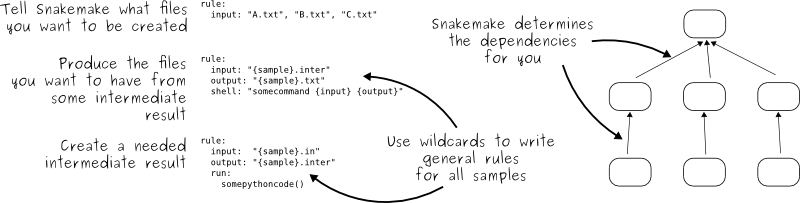
Somebody wrote a Snakemake solution in the Snakefile:
# a list of all the books we are analyzing
DATA = glob_wildcards('data/{book}.txt').book
rule all:
input:
expand('statistics/{book}.data', book=DATA),
expand('plot/{book}.png', book=DATA)
# count words in one of our books
rule count_words:
input:
script='code/count.py',
book='data/{file}.txt'
output: 'statistics/{file}.data'
shell: 'python {input.script} {input.book} > {output}'
# create a plot for each book
rule make_plot:
input:
script='code/plot.py',
book='statistics/{file}.data'
output: 'plot/{file}.png'
shell: 'python {input.script} --data-file {input.book} --plot-file {output}'
We can see that Snakemake uses declarative style:
Snakefiles contain rules that relate targets (output) to dependencies
(input) and commands (shell).
Steps:
Clone the example to your computer:
$ git clone https://github.com/coderefinery/word-count.gitStudy the Snakefile. How does it know what to do first and what to do then?
Try to run it. Since version 5.11 one needs to specify number of cores (or jobs) using
-j,--jobsor--cores:$ snakemake --delete-all-output -j 1 $ snakemake -j 1
The
--delete-all-outputpart makes sure that we remove all generated files before we start.Try running
snakemakeagain and observe that and discuss why it refused to rerun all steps:$ snakemake -j 1 Building DAG of jobs... Nothing to be done (all requested files are present and up to date).
Make a tiny modification to the plot.py script and run
$ snakemake -j 1again and observe how it will only re-run the plot steps.Make a tiny modification to one of the books and run
$ snakemake -j 1again and observe how it only regenerates files for this book.Discuss possible advantages compared to a scripted solution.
Question for R developers: Imagine you want to rewrite the two Python scripts and use R instead. Which lines in the Snakefile would you have to modify so that it uses your R code?
If you make changes to the Snakefile, validate it using
$ snakemake --lint.
Solution
2: Start with “all” and look what it depends on. Now search for rules that have these as output. Look for their inputs and search where they are produced. In other words, search backwards and build a graph of dependencies. This is what Snakemake does.
4: It can see that outputs are newer than inputs. It will only regenerate outputs if they are not there or if the inputs or scripts have changed.
7: It only generates steps and outputs that are missing or outdated. The workflow does not run everything every time. In other words if you notice a problem or update information “half way” in the analysis, it will only re-run what needs to be re-run. Nothing more, nothing less. Another advantage is that it can distribute tasks to multiple cores, off-load work to supercomputers, offers more fine-grained control over environments, and more.
8: Probably only the two lines containing “shell”.
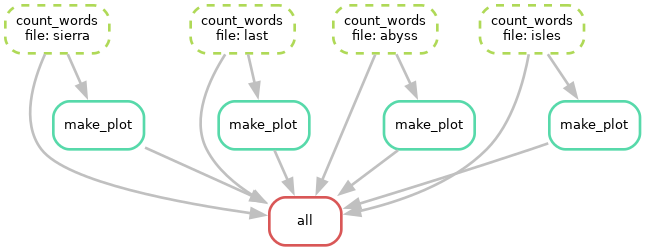
Visualizing the workflow
We can visualize the directed acyclic graph (DAG) of our current Snakefile
using the --dag option, which will output the DAG in dot language.
Note: This requires the Graphviz software,
which can be installed by conda install graphviz.
$ snakemake -j 1 --dag | dot -Tpng > dag.png
Rules that have yet to be completed are indicated with solid outlines, while already completed rules are indicated with dashed outlines.
Why Snakemake?
Gentle learning curve.
Free, open-source, and installs easily via conda or pip.
Cross-platform (Windows, MacOS, Linux) and compatible with all High Performance Computing (HPC) schedulers: same workflow works without modification and scales appropriately whether on a laptop or cluster.
If several workflow steps are independent of each other, and you have multiple cores available, Snakemake can run them in parallel.
It is possible to define isolated software environments per rule, e.g. by adding
conda: 'environment.yml'to a rule.It is also possible to run workflows in Docker or Apptainer containers, e.g. by adding
container: 'docker://some-org/some-tool#2.3.1'to a rule.Heavily used in bioinformatics, but is completely general.
Nice functionality for archiving the workflow, see: the official Snakemake documentation
Tools like Snakemake help us with reproducibility by supporting us with automation, scalability and portability of our workflows.
Similar tools
Many specialized frameworks exist.
Keypoints
Computational steps can be recorded in many ways.
Workflow tools can help if there are many steps to be executed and/or many datasets to be processed.